Band Engineering
For lack of a better name, this Windows program is simply called BandEng.
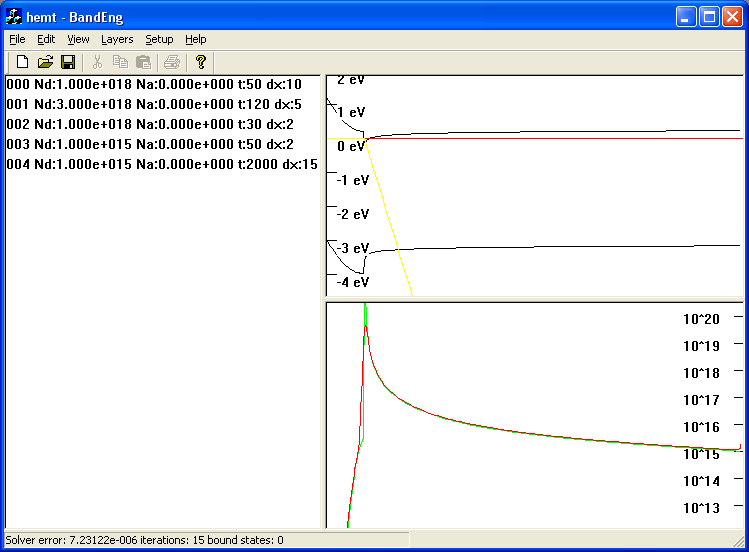
Here is a random (outdated) screenshot of a structure I sort of made up for demonstration purposes:
You can get it here.
A quick tutorial:
The program will have serious problems on some structures with large polarization charges - I'm still working on that. (Update, if it's not converging, i.e. the number of iterations is the same as the max number of iterations, try playing with the setup->solver properties damping factor. .9 usually helps...)
Note:
Note: as of 4/4/04 everything in the materials dialog is used. If you use an older materials file, make sure that the deep donor/acceptor levels are correct for your purposes.
I added CV profiling from the left contact. Look under Layers... You need a "fixed" contact on the left to use this, otherwise it's just going to sit there and whirl away at nothing. The output file is called cv.out.
There's no help with it (for now), but here's the basic rundown:
The program will solve Poisson's and Schrodinger's equations. You can interactively edit the layer stack and view the band diagram on the right side. Polarization is calculated in GaN/AlGaN systems. Currently, there is very limited graphics support, and concentrations are calculated using the Boltzmann approximation - you have been warned (Update 7/5/03 now uses a modified Sommerfeld approximation for degenerate areas.) Also, many menu options simply do not work under the materials database. Feel free to update your database though, because future versions will use those data fields. Also note that the status bar gives kind of bogus information right now. The iterations are right, and that's about it. It's all a work in progress of course...
New functionality (5/17/04): text import
File structure:
material_name x= Nd= Na= Ndd= Nda= relax= Efn= Efp= dx= t=
all above values are optional
Quadratic values are entered in constant,linear,quad terms (no spaces between commas and numbers.)
For example,
x=.2,.4 means that x=.2+.4y where y is the normalized position in the
material (just like in the materials dialog)
you can omit the linear, or linear and quadratic terms if you choose
You can make nested repeated structures using:
repeat=5
(....) body
endrepeat
Here is an example:
GaN t=500 dx=10 Na=5e17
repeat=4
InGaN x=.2 t=50 dx=2
GaN t=50 dx=2 Nd=5e17
endrepeat
GaN t=500 dx=10 Nd=5e17
Have fun!
Please send me an email at mgrundmann at ece dot ucsb dot edu if you use this program. I'm not smart enough to write a script to hide this effectively, so just reconstruct it. I'd really like to know what it's being used for so I can make improvements in the future. If you publish any diagrams that this program creates, please cite it somehow and/or contact me if you'd like a citation example.